
SAĞLIK
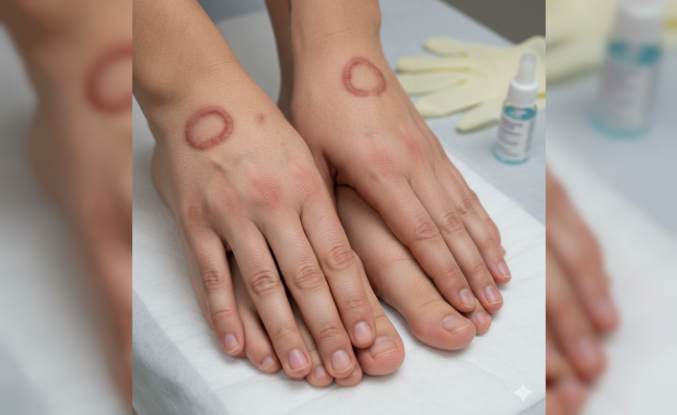


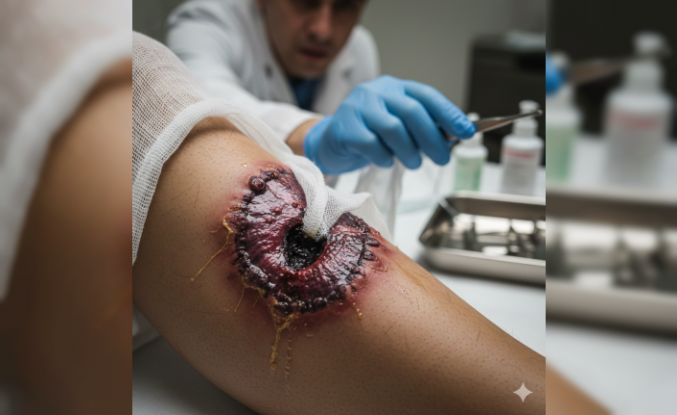


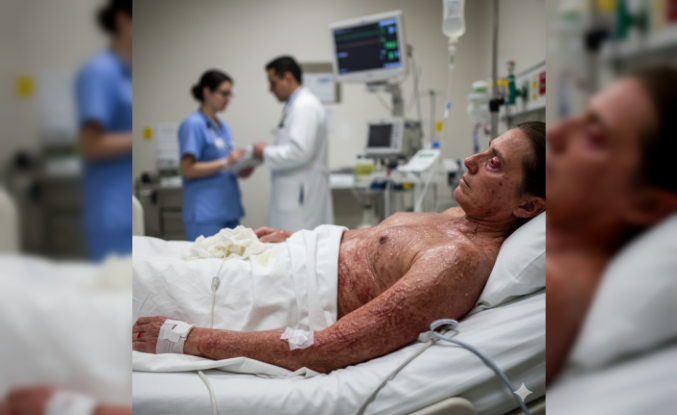


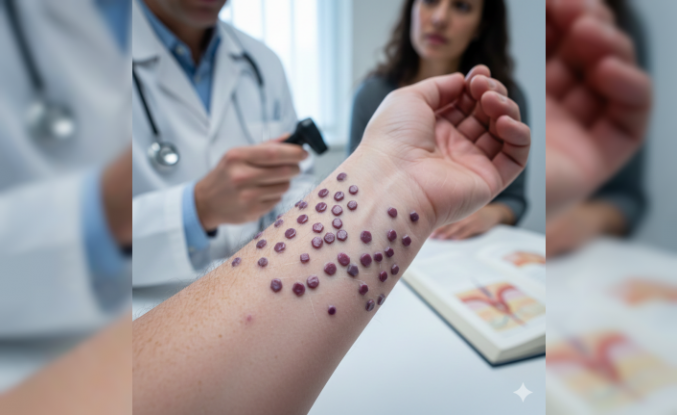


Nadir Görülen Fanconi Sendromu Belirtileri ve Tedavi Yöntemleri
Böbreklerin proksimal tübül adı verilen bölgesinde yaşanan emilim bozukluğu, tıp dünyasında Fanconi sendromu olarak biliniyor. Bu durum vücut için gerekli olan glikoz, aminoasit ve fosfat gibi maddelerin idrarla dışarı atılmasına yol açıyor. Genellikle genetik faktörler nedeniyle ortaya çıkan tablo, bazen ağır metal zehirlenmeleri sonucunda da gelişebiliyor. Böbrek süzme biriminde işler karışmış gibi duruyor. Hastalar halsizlik ve kemik ağrısı şikayetleriyle sağlık kuruluşlarına başvuruyor.

Genetik Tansiyon Hastalığı Lidle Sendromu Mercek Altında
Nadir görülen genetik rahatsızlıklar arasında yer alan Lidle sendromu, özellikle genç yaşlarda ortaya çıkan yüksek tansiyon vakalarıyla dikkat çekiyor. Uzmanlar bu hastalığın böbreklerdeki sodyu (tuz) emilimini kontrol eden kanalların bozulmasıyla oluştuğunu belirtiyor. Vücut aşırı miktarda tuzu tutuyor. Bu durum damarlardaki basıncı artırıyor. Hastane koridorunda bekleyen ailelerin endişesi yüzlerinden okunuyordu.

Nadir Görülen Gitelman Sendromu Takip Altında
Vücudun elektrolit dengesini altüst eden Gitelman sendromu, genetik temelli bir sağlık sorunu olarak son dönemde daha fazla dikkat çekiyor. Böbreklerdeki tübüllerin tuz geri emilimini yapamaması sonucu ortaya çıkan bu durum, halsizlik şikayetiyle başlıyor. Hastalar genellikle ergenlik döneminde belirti gösterse de bazen yetişkinlikte teşhis konulabiliyor.

Nadir Görülen Bartter Sendromu ve Erken Tanının Önemi
Böbreklerdeki tuz geri emilim mekanizmasını bozan Bartter sendromu, özellikle çocukluk çağında ciddi sağlık sorunlarına yol açıyor. Genetik geçişli bu vucut yapısındaki bozukluk, idrarla aşırı miktarda potasyum ve kalsiyum kaybedilmesine neden oluyor. Hastane koridorunda bekleyen ailelerin yüzündeki o yorgun ifade her şeyi özetliyor aslında. Uzmanlar hipokalemi (kan potasyum düşüklüğü) durumunun kas güçsüzlüğüne yol açtığını belirtiyor. Hastalık nadir görülüyor.

Waterhouse Friderichsen Sendromu Nedir Belirtileri Nelerdir
Hastanelerin acil servislerine başvuran vakalar ciddi bir seyir izliyor. Genellikle meningokok (menenjit mikrobu) kaynaklı gelişen bu tablo, sürrenal (böbrek üstü bezi) yetmezliğiyle sonuçlanıyor. Hastalarda ani başlayan yüksek ateş ve döküntüler dikkat çekiyor. Vaka sayısı arttı. Acil servis önündeki kalabalık bekleyiş sürüyor.
Cushing Sendromu Ameliyatı Sonrası Nelson Riski
Böbrek üstü bezleri alınan hastalarda nadir görülen Nelson sendromu takibi hayati önem taşıyor. Uzmanlar cerrahi sonrası ciltteki renk değişimlerine karşı uyarıyor.

Karsinoid Sendrom Vakalarında Artış: Uzmanlar Belirtilere Karşı Uyarıyor
Nöroendokrin tümörlerden kaynaklanan karsinoid sendrom, son dönemde tıp dünyasında daha sık gündeme geliyor. Vücudun hormon üretim dengesini bozan bu durum, genellikle sindirim sistemi veya akciğerlerdeki kitlelerle tetikleniyor. Hastane koridorlarında bekleyen hastaların endişeli bakışları durumun ciddiyetini özetliyor. Kan akışına karışan aşırı serotonin (mutluluk hormonu) miktarı, vücutta beklenmedik tepkimelere yol açmakta.

Nadir Görülen Zollinger Ellison Sendromu Teşhisinde Yeni Yöntemler
Karnın üst bölgesinde geçmeyen şiddetli ağrılarla kendini belli eden Zollinger Ellison sendromu, tıp dünyasının gündeminde yer alıyor. Pankreas veya ince bağırsakta oluşan tümörler sebebiyle ortaya çıkan bu durum, mide asidinin aşırı üretilmesine yol açıyor. Hastanelerin gastroenteroloji (sindirim sistemi birimi) servislerine başvuran pek çok kişi, aslında bu sinsi hastalıkla mücadele ediyor olabilir.

Simmonds Sendromu Belirtileri ve Tanı Süreci
Hipofiz bezinin (beyin tabanındaki ana bez) tam fonksiyon kaybı olarak bilinen Simmonds sendromu tıp dünyasında nadir görülüyor. Hastalık genellikle doğum sonrası ağır kanamalara bağlı gelişen doku ölümüyle ortaya çıkıyor. Belirtiler ilk etapta yorgunluk ve iştahsızlık şeklinde kendisini gösteriyor. Vücut direnci düşüyor.

Everything About Insulin Resistance and Blood Suga
Hello everyone, welcome to MediHaber! In this video, we are journeying into the mysterious world of blood sugar (glucose), our body's most critical energy source. From why you feel tired all day to the real reasons behind sugar cravings; Elif Nur Gezer explains scientific facts that will completely renew your metabolism—from insulin resistance to the correct order of eating. Establishing this "sweet balance," which is the key to a healthy life, is actually much easier than you think. Topics you will find in our video: The magnificent balance between Insulin and Glucagon, Differences between glycemic index and glycemic load, Techniques to slow sugar absorption by changing the order of eating, The massive effects of sleep, stress, and exercise on blood sugar. If you are ready to listen to your body and be the protagonist of your own health, don't forget to watch our video until the end and subscribe to our channel!

Cushing Ameliyatı Sonrası Beklenmedik Misafir: Nelson Sendromu
Nadir görülen bu tablo, aslında bir cerrahi müdahalenin ardından ortaya çıkıyor. Cushing hastalığı nedeniyle iki böbrek üstü bezini de aldıran kişilerde görülmesi muhtemel. Belki de vücudun denge arayışı bu durumu tetikliyor. Uzmanlar, hipofiz bezinin (beyin tabanındaki hormon salgılayan merkez) kontrolsüz büyümesine odaklanmış durumda.

Yüzdeki Kızarıklık Basit Bir Alerji Olmayabilir
Salonda bekleyen hastaların yüzündeki o ani değişim dikkatimi çekiyor. Bir anda parlayan, kızaran ciltler ve hızlanan nefes sesleri. Çoğu kişi bunu heyecana veya yediği bir yemeğe bağlıyor. Oysa mesele çok daha derinlerde, karın içindeki minik bir kitlede saklı duruyor.

Midedeki Sessiz Tehlike: Gastrinoma Alarmı
Hastane koridorlarında bugünlerde farklı bir hareketlilik var. Uzmanlar nadir görülen ama mideyi adeta yakan bir durumu tartışıyor. Zollinger Ellison Sendromu (aşırı asit salgılanması) teşhisi konulan hastaların sayısı artıyor mu sorusu akıllarda. Aslında mesele sadece bir mide ağrısı değil.

Doğum Mucizesinin Gölgesindeki Gizli Tehlike: Sheehan Sendromu
Hastane koridorlarında sevinç çığlıkları yükselirken bazen sessiz bir fırtına kopuyor. Doğum sırasında yaşanan şiddetli kanamalar, yeni annelerin hayatını hiç beklenmedik şekilde karartabiliyor. Uzmanlar uyarıyor: doğum sonu (doğum sonrası) süreçte geçmeyen halsizlik basit bir yorgunluk olmayabilir.

Görünmez Tehlike: Hipofiz Bezi Neden Susar?
Geçenlerde bir klinik koridorunda beklerken rastladım bu sessiz mücadeleye. Şimonds hastalığı (hipofiz yetmezliği) aslında vücudun ana kumanda merkezinin aniden kepenk indirmesi gibi bir durum. Her şey normal giderken hormonların çekilmesiyle insan bir anda tükeniyor. Zor.

Sessiz Tehlike: Vücudun Enerji Santrali Durduğunda
Hastanenin acil servis koridorunda bekliyoruz, içeride bir yaşam mücadelesi var. Tiroid bezinin artık çalışmadığı, vücudun adeta donduğu o an yaşanıyor. Myxedema (miksedem) koması, aslında çok nadir görülen ama oldukça ağır bir tablo. İnsan şaşırıyor, nasıl bu noktaya gelinir diye. Her şey bazen basit bir halsizlikle başlıyor.

Gözlerdeki Gizemli Büyüme: Graves Tehlikesi
Hastanelerin endokrinoloji koridorlarında son dönemde bir hareketlilik göze çarpıyor. İnsanlar ellerindeki titremeden veya aynaya baktıklarında gördükleri o yabancı bakıştan şikayetçi. Graves hastalığı tam olarak burada başlıyor: bağışıklık sisteminin kendi tiroidine saldırması. Vücut adeta kendi kendine savaş açıyor. Garip.








