
SAĞLIK














Ciltte Bitmeyen Kaşıntı: Atopik Dermatit Artışta
Hastanelerin dermatoloji kliniklerinde son dönemde yoğunluk oluşturan şikayetlerin başında kronik cilt kurulukları geliyor. Halk arasında alerjik egzama olarak bilinen atopik dermatit vakaları, özellikle mevsim geçişlerinde yetişkin ve çocuklarda ciddi rahatsızlıklara yol açıyor. Uzmanlar bu durumun sadece genetik değil, çevresel faktörlerle de doğrudan bağlantılı olduğunu her fırsatta dile getiriyor.

Gözlerdeki Bulanık Görüşün Sebebi Astigmatizma Olabilir
Göz merceğinin veya korneanın düzensiz kavislenmesi sonucu ortaya çıkan astigmatizma, günümüzde toplumun büyük kesimini etkiliyor. Işığın retinaya odaklanırken tek bir noktada toplanamaması durumu, bireylerde hem uzak hem de yakın görüşte ciddi sorunlar yaratmaktadır. Yapılan son saha araştırmalarına göre nüfusun yaklaşık %30'u bu sorundan muzdaripken, klinik veriler bu oranı bazen %32 olarak kaydediyor.

Sanayi Bölgelerinde Asit Yağmuru Alarmı: Ekipler Teyakkuzda
Hava kirliliğinin yoğunlaştığı bölgelerde son dönemde artan emisyon değerleri, asit yağmuru riskini yeniden gündeme taşıdı. Meteoroloji uzmanları özellikle sanayi havzalarında yağışların kimyasal yapısının değişebileceği konusunda ciddi uyarılarda bulunuyor. Fabrika bacalarından çıkan gazlar, bulutlardaki su buharıyla birleşince ortaya çıkan bu durum doğayı tehdit ediyor.

Eklemlerdeki Gizli Tehlike: Artrit Vakaları Artış Gösteriyor
Dünya genelinde milyonlarca insanı etkileyen eklem iltihaplanmaları, son dönemde sağlık gündeminin üst sıralarına tırmandı. Tıbbi adı artrit (eklem iltihabı) olan bu rahatsızlık, sadece yaşlıları değil artık genç nüfusu da tehdit ediyor. Hastanelerin romatoloji polikliniklerine başvuran hasta sayısında geçtiğimiz yıla oranla yaklaşık %22 civarında bir artış gözlemleniyor. Uzmanlar ise bu durumu hareketsiz yaşam tarzı ve beslenme alışkanlıklarına bağlıyor.

Günlük Yaşamı Zorlaştıran Gizli Engel: Apraksi
Beynin kas hareketlerini kontrol etme yeteneğini etkileyen apraksi, son yıllarda nörolojik vakalar arasında daha sık görülmeye başlandı. Kişinin fiziksel gücü yerinde olmasına rağmen, beynin yapmak istediği eylemi kaslara doğru iletememesi durumu olan bu rahatsızlık yaşam kalitesini ciddi oranda düşürüyor. Uzmanlar özellikle çocukluk çağı konuşma apraksisi (sözel planlama bozukluğu) tanısında artış olduğunu belirtiyor.

Akne Sorunu Sosyal Yaşamı Ve Cilt Sağlığını Tehdit Ediyor
Cilt yüzeyinde oluşan sivilceler toplumun genelinde sadece bir ergenlık sorunu olarak görülse de uzmanlar durumun tıbbi boyutuna dikkat çekiyor. Son yapılan saha araştırmalarına göre yetişkinlerin %82'lik bir kısmında dönemsel akne problemleri gözlemlenirken, kronik vakaların oranı %18 civarında seyretmeye devam ediyor. Hastanelerin dermatoloji polikliniklerinde sabah saatlerinden itibaren yoğunluk yaşanırken doktorlar randevu yetiştirmekte zorlanıyor. Koridorda bekleyen hastaların yüzündeki bıkkınlık durumu özetliyor gibiydi.

Şehir Hayatında Artan Kaygı: Anksiyete Bozukluğu Mercek Altında
Modern yaşamın getirdiği hız ve belirsizlik, toplum genelinde piskolojik yükleri artırmaya devam ediyor. Yapılan son araştırmalara göre Türkiye genelinde her on kişiden dördü hayatının bir döneminde yoğun kaygı hissediyor. Uzmanlar bu durumun klinik bir tabloya dönüşmesi durumunda anksiyete bozukluğu (kaygı bozukluğu) tanısının konulduğunu belirtiyor. Bu rahatsızlık sadece zihinsel bir süreç değil, vücudun genel işleyişini de doğrudan etkileyen bir süreçtir.

Toplumun Görünmez Yüzü: Antisosyal Kişilik Bozukluğu Vakaları Artıyor mu?
Sokakta yanınızdan geçen veya iş yerinde aynı masayı paylaştığınız birinin duygusal dünyasındaki boşluğu fark etmek her zaman kolay olmuyor. Uzmanlar halk arasında psikopati (duygusuzluk hali) olarak da bilinen antisosyal kişilik bozukluğu tanısı konulan bireylerin sayısında son yıllarda belirgin bir değişim gözlemlendiğini belirtiyor. Özellikle sosyal kurallara uyum sağlamakta zorlanan bu kişiler, empati (duygudaşlık) yeteneklerinin kısıtlı olmasıyla dikkat çekiyor. Pisikiyatrist kliniklerinde yapılan gözlemler bu tablonun sadece bireysel değil toplumsal bir mesele olduğunu kanıtlar nitelikte.

Koku Duyusu Kaybı Hayat Kalitesini Etkiliyor
Burnun en temel işlevi olan koku alma yeteneğinin tamamen yitirilmesi olarak bilinen anosmi, son yıllarda toplumda daha sık görülmeye başlandı. Uzmanlar bu durumun sadece fiziksel değil, psikolojik süreçleri de tetiklediğini belirtiyor. Aniden gelişen koku kaybı (anosmi) vakalarında hastaların çoğu durumu ilk başta geçici bir soğuk algınlığı zannediyor. Ancak haftalar süren tıkanıklık sonrası koku alamama hali kalıcı hasarın habercisi olabiliyor.

Modern Çağın Sessiz Tehlikesi Anoreksiya Nervoza Mercek Altında
Uzmanlar son yıllarda genç popülasyonda hızla artan yeme bozukluklarına karşı önemli uyarılarda bulunuyor. Özellikle ergenlik dönemindeki bireylerde görülen anoreksiya nervoza (aşırı zayıflık takıntısı) vakaları, fiziksel sağlık kadar ruh sağlığını da ciddi biçimde tehdit etmekte. Yapılan saha araştırmaları, sosyal medya kullanımının beden algısı üzerindeki olumsuz etkilerini açıkça ortaya koyuyor. Hastanenin koridorunda bekleyen ailelerin yüzündeki o belirsiz ifade durumu özetliyor aslında.

Ege Denizi'nde Korkutan Sarsıntı: Ekipler Teyakkuzda
Ege Denizi açıklarında sabah saatlerinde meydana gelen deprem, kıyı şeridindeki yerleşim birimlerinde kısa süreli paniğe neden oldu. Afet ve Acil Durum Yönetimi Başkanlığı (AFAD) tarafından yapılan açıklamaya göre, sarsıntının merkez üssü deniz tabanının yaklaşık 12 kilometre derinliğinde gerçekleşti. Bölgedeki sismik (depremle ilgili) hareketlilik, yerel saatle 09.45 sularında kaydedildi.

Gözde İris Yokluğu: Aniridi Hakkında Bilinmesi Gerekenler
Dünya genelinde oldukça nadir rastlanan ve genellikle doğumla birlikte gelen aniridi (gözün renkli kısmının yokluğu) vakaları tıp dünyasının radarında kalmaya devam ediyor. Görme yetisini ciddi oranda kısıtlayan bu durum, sadece bir estetik sorun değil, aynı zamanda çok boyutlu bir sağlık problemidir. Gözün ışığı süzme görevini üstlenen iris tabakasının tam veya kısmi eksikliği, hastaların ışığa karşı aşırı duyarlı olmasına yol açıyor.

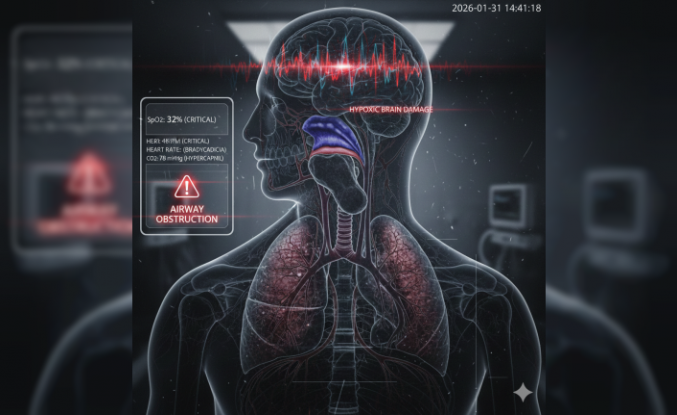
Sessiz Tehlike Anevrizma: Belirtiler ve Risk Faktörleri
Damar duvarının zayıflaması sonucu oluşan anevrizma, toplumda "sessiz katil" olarak biliniyor. Genellikle hiçbir ön belirti vermeden ilerleyen bu durum damarın bir bölgesinde balonlaşma (damar genişlemesi) meydana getiriyor. Sağlık Bilimleri Üniversitesi uzmanları her yıl binlerce kişinin bu sinsi rahatsızlık sebebiyle acil servislere başvurduğunu belirtiyor. Türkiye genelinde görülme sıklığı yüzde 2 ile 3 civarında seyrederken vakaların çoğu tesadüfen saptanıyor.

Amnazi Bölgesinde Altyapı Çalışmaları Hız Kazandı
Bölge halkının uzun süredir beklediği modern kanalizasyon ve yağmur suyu hattı projesinde ekipler vites yükseltti. Belediye ekipleri, sabahın erken saatlerinde Amnazi ana arteri üzerinde kazı işlemlerine başlarken trafik akışı geçici güzerga üzerinden sağlanıyor. Proje kapsamında yaklaşık 850 metre uzunluğunda yeni bir boru hattı döşenmesi planlanıyor. Yetkililer, kış ayları bastırmadan bu çalışmanın tamamlanacağını belirtti.

Kadın Sağlığında Görülmeyen Engel: Amenore ve Güncel Tanı Yöntemleri
Kadın sağlığını doğrudan etkileyen ve halk arasında adet görememe olarak bilinen Amenore (regl yokluğu), modern tıp dünyasında geniş bir yer tutuyor. Uzmanlar bu durumu primer ve sekonder olmak üzere iki ana başlık altında incelerken, vakaların artış göstermesi dikkat çekiyor. Ergenlik döneminde 15 yaşına kadar hiç kanama olmaması durumu birincil tip olarak adlandırılırken, düzenli döngüsü olan bir kadının en az üç ay boyunca adet görmemesi ikincil tip sayılıyor.

Çocuklarda Göz Tembelliği Riski: Erken Teşhis Hayati Önem Taşıyor
Halk arasında göz tembelliği olarak bilinen ambliyopi, çocukluk çağında görme gelişiminin normal seyretmemesi sonucu ortaya çıkıyor. Uzmanlar özellikle okul öncesi dönemdeki çocukların rutin kontrollerinin aksatılmaması gerektiği konusunda aileleri uyarıyor. Görsel sistemin tam kapasiteyle çalışabilmesi için her iki gözden de beyne net sinyal gitmesi gerekiyor.








