Tricorhinofalangeal Sendromu (TRPS): Kıl, Burun ve Parmakları Etkileyen Nadir Genetik Durumun Tıbbi Yolculuğu
Kıl, Burun, Parmaklar: Gizemli Sendromun İzleri
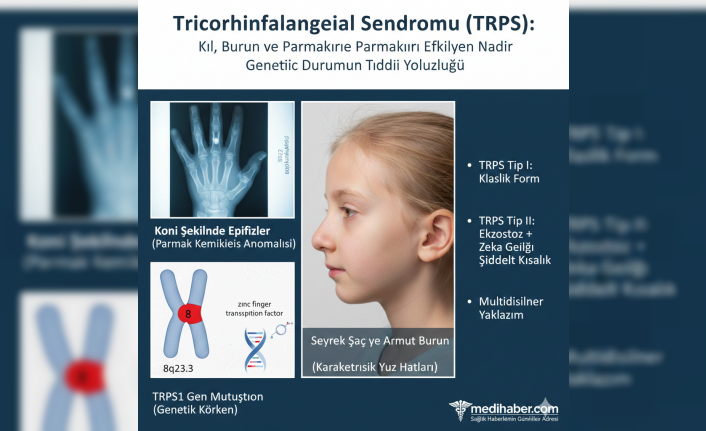
Trikorhinofalangeal sendrom (TRPS), adını etkilendiği temel vücut bölgelerinden alan nadir bir kalıtsal hastalıktır: Tricho (kıl), rhino (burun) ve falangeal (parmaklar). Bu genetik bozukluk, bireylerde belirgin fiziksel özelliklere ve iskelet anomalilerine yol açar. Tıbbi literatürde ilk olarak 1966'da Dr. Giedion tarafından tanımlanan bu durum, uzun yıllar boyunca sadece klinik gözlemlere dayanılarak teşhis edildi. Bugün ise modern genetik bilimi sayesinde sendromun kökeni ve çeşitleri hakkında çok daha net bilgilere sahibiz.
Tarihsel Dönüm Noktaları ve Doğrulanmış Bilgiler
Tricorhinofalangeal sendromun anlaşılmasındaki en büyük sıçrama, genetik temelin keşfiyle gerçekleşti. Sendromun temelini oluşturan TRPS1 geni, 2000 yılında haritalandı ve 8. kromozomda (8q23.3) yer aldığı tespit edildi. Bu gen, çinko parmak transkripsiyon faktörü (zinc finger transcription factor) adı verilen ve kemik, kıkırdak ve saç foliküllerinin gelişimini düzenleyen bir proteini kodlar. TRPS1 genindeki mutasyonlar (değişiklikler) bu proteinin işlevini bozarak sendromun karakteristik belirtilerine yol açıyor.
Sendromun üç ana tipi tanımlanmıştır ve bunlar arasındaki farklar bilimsel yayınlarla doğrulanmıştır:
-
TRPS Tip I: En sık görülen, klasik formdur ve TRPS1 genindeki patojenik varyantlardan (hastalığa neden olan genetik değişimler) kaynaklanır. Seyrek saç, armut biçimli burun ucu ve parmak kemiklerinde koni şeklinde epifizler (kemiğin büyüme ucu) temel bulgulardır.
-
TRPS Tip II (Langer-Giedion Sendromu): Bu, bitişik gen silinme sendromudur (contiguous gene deletion syndrome); yani sadece TRPS1 geni değil, aynı zamanda EXT1 geni gibi diğer bazı komşu genler de silinmiştir. Bu durum, Tip I belirtilerine ek olarak genellikle multiple ekzostozlar (çok sayıda iyi huylu kemik çıkıntıları) ve zihinsel gerilik (mental retardasyon) ile seyreder.
-
TRPS Tip III: En nadir görülen tiptir. Tip I'e benzer ancak tüm falanks (parmak kemiği) ve metakarplardaki (el tarağı kemiği) aşırı kısalık (şiddetli brakidaktili) ile karakterizedir.
Bu tiplendirme ve genetik korelasyonlar, uzman doktorların ve akademik kaynakların fikir birliğiyle kabul ettiği doğrulanmış tıbbi bilgilerdir.
Sendromun Klinik Tablosu ve Yanıltıcı İddialar
Sendromun fiziksel bulguları oldukça ayırt edicidir, ancak bireyler arasında değişken ekspresivite (belirti şiddetinin farklı olması) göstermesi mümkündür.
Doğrulanmış Klinik Bulgular:
Shutterstock
Keşfet
-
Kıl (Tricho): Saçlar genellikle ince, seyrek (özellikle şakaklarda ve yanlarda) ve yavaş uzayan yapıdadır. Kaşların yan kısımları da seyrek olabilir.
-
Burun (Rhino): Burun, soğan biçimli (bulböz) uca sahip, geniş sırtlı ve burun kanatları (ala nasi) gelişmemiş görünümdedir. Üst dudak ile burun arasındaki dikey oluk (filtrum) uzun ve düz olabilir.
-
Parmaklar (Phalangeal): El ve ayak parmaklarında kısa parmaklılık (brakidaktili) ve radyolojik incelemelerde görülen koni şeklinde epifizler (bu, büyüme plağının erken kapanmasına neden olabilir) en önemli iskelet bulgularıdır.
-
Diğer İskelet Sorunları: Kısa boy, erken başlayan kalça eklemi sorunları (Perthes benzeri bulgular veya kalça displazisi) ve eklem hipermobilitesi (aşırı hareketlilik) de sıklıkla gözlenir. Erken tanı, özellikle kalça gibi büyük eklemlerdeki sorunların yönetimi için kritik öneme sahiptir.
Haber Metnindeki Mesaj ve Doğruluk/Yanlışlık Değerlendirmesi
Haber metnindeki mesaj, muhtemelen "nadiren görülen bu genetik durumun belirtilerinin tanınması, erken teşhis ve daha iyi izlemeyi sağlar" şeklindedir. Bu mesaj, modern tıbbın ve akademik çalışmaların tamamen doğruladığı bir gerçektir. Dismorfik özelliklerin (kendine has fiziksel görünüm) tanınması, özellikle çocuk doktorları için önemlidir çünkü bu hastaların yaşam kalitesini artıracak ortopedik ve diğer tedavilerin zamanında başlamasını sağlar.
Hastalık hakkında dolaşan bazı yanlış veya eksik bilgiler olabilir. Örneğin, tüm tiplerde zeka geriliği olduğu iddiası yanlıştır. Zihinsel gerilik, yalnızca EXT1 geni silinmesinin de görüldüğü TRPS Tip II'ye özgü bir bulgudur; TRPS Tip I ve Tip III'te zeka genellikle normaldir.
Klinik Uygulamalar ve Gelecek Perspektifi
Trikorhinofalangeal sendromun teşhisi, artık sadece fiziksel muayene ve radyolojik görüntüleme (el ve ayak grafileri) ile değil, kesin tanı için genetik testlerle (TRPS1 gen analizi) konulmaktadır. Sendromun yönetimi ise multidisipliner bir yaklaşım gerektirir: ortopedistler iskelet anormalliklerini, dermatologlar saç ve tırnak sorunlarını, diş hekimleri de dental anomalileri tedavi eder. TRPS’de erken tanı yaşam kalitesini artırır ve kronik eklem sorunlarının ilerlemesini yavaşlatabilir.
Hastalığın prevalansı (yaygınlığı) henüz kesin olarak bilinmese de, dünya genelinde birkaç yüz vaka bildirilmiştir. Klinik gözlemler gösteriyor ki, belirtileri hafif olan pek çok birey fark edilmeden kalabilmekte ve bu durum, TRPS'nin nadir bir genetik hastalık olmasından kaynaklanmaktadır. Bu genetik rahatsızlığın kökenine inen moleküler çalışmalar hız kesmeden devam ediyor. Gen terapileri gibi yenilikçi tedavi yaklaşımları henüz klinik aşamaya gelmemiş olsa da, TRPS1 geni fonksiyonu araştırmaları gelecekteki hedefli tedaviler için umut verici bir zemin hazırlıyor. Gelişmeleri medihaber.com'u takip ve sosyal medya hesaplarını takip ederek güncel bilgilere ulaşabilirsiniz. Uzmanlar, bu nadir hastalıkla yaşayan kişilerin tıbbi ihtiyaçlarına yönelik daha kişiselleştirilmiş tedavi protokollerinin geliştirilmesi için çabalıyor.
Son Gelişmeler Işığında Uzman Görüşleri
Son dönemde yayımlanan akademik çalışmalar, özellikle TRPS Tip I hastalarındaki erken başlangıçlı osteoartrit (eklem kireçlenmesi) ve kalça eklemi sorunlarının önemini vurgulamakta. Uzmanlar, büyüme çağındaki hastaların düzenli ortopedik takibinin ve fizik tedavisinin, ilerideki hareket kısıtlılıklarını minimize etmede önemli bir rol oynadığını belirtiyor. Hastalığın otozomal dominant (baskın) kalıtım şekli, ebeveynlerden birinde mutasyon varsa çocuğa geçme riskinin %50 olduğu anlamına geliyor; bu da genetik danışmanlığın önemi konusunu gündeme taşıyor.
Bu genetik durumun farkındalığını artırmak ve tanı süreçlerini hızlandırmak, sadece hastaların değil, aynı zamanda ailelerinin de yaşamlarını iyileştirecek en önemli adımlardan biridir. Tıbbi kaynaklar, bu sendromun sadece bir dizi fiziksel özellikten ibaret olmadığını, aynı zamanda yönetilmesi gereken ciddi iskelet ve eklem sorunlarını da beraberinde getirdiğini net bir şekilde ortaya koyuyor.